The ultimate BLAST platform for genes
Kent Informatics, Inc. is a small bioinformatics software company based in Santa Cruz, California. We provide scientists with tools to analyze the human genome, particularly, BLAT - The BLAST-like Alignment Tool. Our clients include pharmaceuticals, genomics, and bioinformatics companies. Our software is always free for universities and non-profit institutions.
GenomeBLAT: A Technical Overview
GenomeBLAT, a specialized bioinformatics platform, utilizes the BLAST-like Alignment Tool (BLAT), designed for ultra-fast sequence alignment. Unlike traditional BLAST, BLAT is optimized for aligning nucleotide or protein sequences across large genomic datasets with higher speed. Its efficiency arises from how it builds indices for genomes (DNA/mRNA) and searches for homologies in large biological sequences.
Core Functions of GenomeBLAT
- BLAT (BLAST-Like Alignment Tool): BLAT is the flagship tool of GenomeBLAT and is particularly advantageous in sequence searching where rapid alignment across expansive data is needed. BLAT works by creating an index of the genome (the reference database) and using this index to quickly identify regions of interest, such as homologies in genomic, mRNA, or protein sequences. It is often used for tasks like identifying gene duplications, orthologous regions, or sequence variations. Because it is highly optimized for speed, BLAT is favored in scenarios where large-scale genomic queries must be handled promptly.
- In-silico PCR: This feature allows users to simulate PCR amplifications, searching for specific sequences within a genome using primer sequences. In-silico PCR offers a fast and reliable way to predict PCR products, aiding in experimental design and validation by ensuring the correct target region is amplified in silico before proceeding with actual lab experiments.
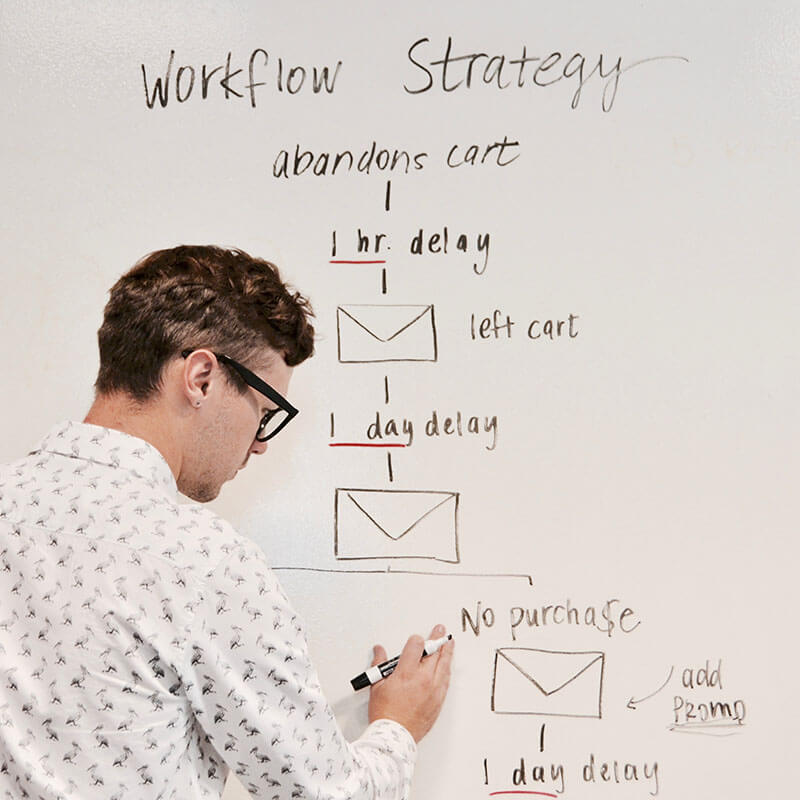
- Genome Browser Integration: GenomeBLAT often integrates with genome browsers, allowing seamless visualization of alignment results. The results from BLAT can be visualized in reference to known gene models, regulatory regions, or other genomic annotations, providing a comprehensive overview of how sequences align within the genome structure. This integration is especially useful in exploring large-scale genomic variations, synteny, and conserved regions across species.
- Applications in Comparative Genomics: GenomeBLAT is highly effective in comparative genomics, allowing researchers to study evolutionary relationships between species by aligning orthologous regions. This is particularly useful in identifying conserved sequences or regions under evolutionary pressure across species, often helping pinpoint functional regions such as genes, promoters, or enhancers.
- Clinical Genomics and Diagnostics: BLAT's speed and accuracy also find applications in clinical genomics, where quick alignment of patient-derived genomic sequences against reference databases is critical for diagnostics. GenomeBLAT can aid in identifying disease-causing mutations, structural variants, and novel sequence variations. Its ability to handle vast genomic datasets while delivering fast results makes it a practical tool in clinical settings.
Performance and Advantages Over BLAST
BLAT's main advantage over BLAST lies in its speed. While BLAST searches by comparing sequences directly, BLAT creates an index of the sequences and uses this index to efficiently retrieve matches. This approach makes BLAT well-suited for large-scale genomic data analysis where rapid results are crucial. BLAT typically excels in situations where:
- Aligning long nucleotide sequences (mRNA/DNA) to large reference genomes.
- Handling repetitive sequences or highly duplicated genes.
- Working in real-time clinical or bioinformatic workflows where time is a limiting factor.
BLAT is less suited for short query sequences or smaller, more precise alignments where BLAST may offer better accuracy at the cost of speed. However, for large-scale genomic studies, BLAT's performance is unmatched in terms of computational efficiency.
Future Prospects and Enhancements
GenomeBLAT continues to evolve, incorporating new algorithmic improvements and better integration with other bioinformatics tools and databases. The ongoing improvements focus on increasing the scalability of the platform to handle ever-expanding genomic datasets, especially as sequencing technologies continue to improve. The future of GenomeBLAT may also see the inclusion of more cloud-based features, allowing even faster and more distributed genomic searches across global research communities.
GenomeBLAT's powerful suite of tools remains indispensable for researchers involved in genomics, evolutionary biology, and bioinformatics, providing efficient and reliable sequence alignment solutions in an increasingly data-heavy world.
What we can do, for you
Digital Transformation
Genome BLAT
Faster. More Accurate. That Simple.
BLAT is a bioinformatics software a tool which performs rapid mRNA/DNA and cross-species protein alignments. BLAT is more accurate and 500 times faster than popular existing tools for mRNA/DNA alignments and 50 times faster for protein alignments at sensitivity settings typically used when comparing vertebrate sequences. (Source: Kent, W.J. 2002. BLAT -- The BLAST-Like Alignment Tool. Genome Research 4: 656-664.)
BLAT is not BLAST. DNA BLAT works by keeping an index of the entire genome (but not the genome itself) in memory. Since the index takes up a bit less than a gigabyte of RAM, BLAT can deliver high performance on a reasonably priced Linux box.
BLAST Farm Headaches Got You Down?
BLAST farm thrashing and crashing? Nodes down and out of sync? Systems administrators frazzled and grumpy? Still waiting after a month for all of those ESTs to align and now there's a new genome assembly? Server hard disk filled yet again?
BLAT will do in a day what BLAST will do in a month, and in many cases a single CPU will handle the load. The output is small and easy to parse. Splice sites are found without exon bleed-over. Give your staff and your server a rest and switch to BLAT today.
Please contact us for licensing information, installation instructions, and a thirty-day free trial.
In-Silico PCR
This bioinformatics tool searches a sequence database with a pair of PCR primers. It uses an indexing strategy to do this quickly. When the search is successful the output is a fasta format sequence file containing all the regions in the database that lie between the primer pair.
You can try out In-Silico PCR interactively.
Please contact us for licensing information, installation instructions, and a thirty-day free trial.
The UCSC Genome Browser
Kent Informatics, Inc. does not own the UCSC Genome Browser. This popular research tool is available for commercial license directly from UCSC.

Are you a startup or a seasoned company looking for a new brand identity?
Are you an established brand looking for ongoing creative services?
About the UCSC Genome Bioinformatics Site
This site contains the reference sequence and working draft assemblies for a large collection of genomes. It also provides a portal to the ENCODE project.
We encourage you to explore these sequences with our tools. The Genome Browser zooms and scrolls over chromosomes, showing the work of annotators worldwide. The Gene Sorter shows expression, homology and other information on groups of genes that can be related in many ways. Blat quickly maps your sequence to the genome. The Table Browser provides convenient access to the underlying database. VisiGene lets you browse through a large collection of in situ mouse and frog images to examine expression patterns.
The UCSC Genome Browser now includes the latest draft assembly of the opossum genome. The Jan. 2006 release of Monodelphis domestica (UCSC version monDom4) was sequenced and assembled by The Broad Institute, Cambridge, MA, USA.
This draft, which has approximately 6.5X coverage, has an assembly length of nearly 3.61 billion bp including gaps (3.50 billion bp without gaps) contained on chromosomes 1-8, X, and Un. The N50 of the genome including gaps is 104,359 bp; the N50 without gaps is 107,990. The N50 size is the length such that 50% of the assembled genome lies in blocks of the N50 size or longer.
The monDom4 sequence and annotation data can be downloaded from the Genome Browser FTP server or Downloads page. Please review the guidelines for using the opposum assembly data.
Many thanks to The Broad Institute for providing these data. The UCSC opossum Genome Browser was produced by Hiram Clawson, Archana Thakkapallayil, Ann Zweig, Kayla Smith and Donna Karolchik. The initial set of annotation tracks was generated by the UCSC Genome Bioinformatics Group. See the Genome Browser Credits page for a detailed list of the organizations and individuals who contributed to the release of this browser.
Conditions of Use
The sequence and annotation data displayed in the Genome Browser are freely available for academic, nonprofit, and personal use with the following conditions:
Genome sequence data use restrictions are noted within the species sections on the Credits page.
Some annotation tracks contributed by external collaborators contain proprietary data that have specific use restrictions. To check for restrictions associated with a particular genome assembly, review the database/README.txt file in the assembly's downloads directory.
The Genome Browser and Blat software are free for academic, nonprofit, and personal use. A license is required for commercial use. See the Licenses page for more information.
Program-driven use of this software is limited to a maximum of one hit every 15 seconds and no more than 5,000 hits per day.
For assistance with questions or problems regarding the UCSC Genome Browser software, database, genome assemblies, or release cycles, see the FAQ.